

VSbet
M88
Fun88
VN88
FB88
BK8
W88
188BET
New88
i9BET
- BK8 – Nhà cái châu Âu #1 trong lòng các cược thủ
- 12play – Nhà cái châu Âu được bet thủ đánh giá uy tín nhất thị trường
- We88 – Thưởng ngay 100K tiền cược miễn phí khi xác nhận tài khoản
- B9Casino – Miễn phí săn thẻ tín dụng SGD32
- 1xBet – Nhà cái châu Âu hỗ trợ cá cược bằng tiền điện tử trên website và App mobile
- Instant Casino – Đăng ký tài khoản nhận thưởng chào mừng tới €7.500
- CoinCasino – Nhà cái cá độ bóng đá bằng tiền điện tử chất nhất
- SambaSlot – Tiền thưởng chào mừng 200% + 50 vòng quay miễn phí
- GoldenPanda – Ưu đãi chào mừng 200% lên đến $7,500 + Hoàn tiền 10% hàng tuần
- InstaSpin – Gói chào mừng lên đến 26.000.000 VND + 100 vòng quay miễn phí
Trên thị trường hiện nay không chỉ xuất hiện các nhà cái có nguồn gốc từ châu Á mà còn có không ít các nhà cái đến từ châu Âu để người chơi lựa chọn tham gia cá cược. Nếu bạn đang băn khoăn không biết đâu là những nhà cái châu Âu uy tín và đáng tin cậy nhất thì hãy cùng tham khảo danh sách dưới đây!
Các nhà cái đến từ châu Âu tốt nhất hiện nay
- BK8 – Nhà cái châu Âu #1 trong lòng các cược thủ
- 12play – Nhà cái châu Âu được bet thủ đánh giá uy tín nhất thị trường
- We88 – Thưởng ngay 100K tiền cược miễn phí khi xác nhận tài khoản
- B9Casino – Miễn phí săn thẻ tín dụng SGD32
- 1xBet – Nhà cái châu Âu hỗ trợ cá cược bằng tiền điện tử trên website và App mobile
- Instant Casino – Đăng ký tài khoản nhận thưởng chào mừng tới €7.500
- CoinCasino – Nhà cái cá độ bóng đá bằng tiền điện tử chất nhất
- SambaSlot – Tiền thưởng chào mừng 200% + 50 vòng quay miễn phí
- GoldenPanda – Ưu đãi chào mừng 200% lên đến $7,500 + Hoàn tiền 10% hàng tuần
- InstaSpin – Gói chào mừng lên đến 26.000.000 VND + 100 vòng quay miễn phí
Đánh giá tổng quan về các nhà cái đến từ châu Âu
BK8 – Nhà cái châu Âu #1 trong lòng các cược thủ
Nhắc tới các nhà cái đến từ châu Âu uy tín nhất hiện nay chắc chắn phải kể tới BK8. Đi vào hoạt động từ năm 2015, tới nay, BK8 đã thành công chinh phục được cộng đồng bet thủ châu Âu và cả châu Á. Số lượng thành viên đăng ký tại BK8 đã lên tới con số hàng chục triệu. Đó là bởi nhà cái luôn mang tới cho người chơi những sản phẩm cá cược chất lượng như:
BK8 – Nhà cái châu Âu #1 trong lòng các cược thủ
Cá cược thể thao, cá cược Esport, Slot game, Sòng bài trực tuyến, Bắn cá,… với tỷ lệ thưởng cao, thanh toán tiền thưởng nhanh chóng, sòng phẳng.
 Tên công ty Tên công ty |
Godric Investments Limited |
 Địa chỉ Địa chỉ |
Số 02, Spinola Road, St. Julians STJ 3014, Malta |
 Giấy phép Giấy phép |
Gaming Curacao |
 Chương trình VIP Chương trình VIP |
Có |
 Ứng dụng Ứng dụng |
Có |
 Phí Phí |
Không |
 Ưu điểm:
Ưu điểm:
- Nhà cái châu Âu hợp pháp, được chính phủ Curacao cấp giấy phép hoạt động
- Có cả website và App mobile để người chơi cá cược dễ dàng
- Kho game cá cược đồ sộ, tới từ các nhà phát hành game uy tín
- Hỗ trợ cá cược bằng cả tiền fiat và crypto
 Nhược điểm:
Nhược điểm:
- Chưa đa dạng các chương trình khuyến mãi
12play – Nhà cái châu Âu được bet thủ đánh giá uy tín nhất thị trường
Một trong những nhà cái châu Âu đang hoạt động tích cực nhất ở nước ta phải kể tới 12play. Nhà cái là một sân chơi cung cấp dịch vụ cá cược online hoàn toàn hợp pháp và có tiềm lực tài chính mạnh.

12play – Nhà cái châu Âu được bet thủ đánh giá uy tín nhất thị trường
Hiện 12play đang hợp tác cùng nhiều nhà phát hành game lớn để cung cấp cho người chơi những tựa game cá cược hấp dẫn với đa dạng kiểu cược thuộc thể loại Bắn Cá, Nổ Hũ, Xổ Số, Sòng Bài, cá cược thể thao,… Các trò chơi do 12play cung cấp đều có tỷ lệ ăn thưởng cao và được thanh toán tiền thưởng đầy đủ ngay sau khi có kết quả cược.
| Tên công ty |
12Play |
| Địa chỉ |
Singapore |
| Giấy phép |
PAGCOR |
| Chương trình VIP |
Có |
| Ứng dụng |
Có |
| Phí |
Không |
Ưu điểm:
- Trang cá cược hợp pháp, được chính phủ Philippines cấp phép kinh doanh
- Kho game cá cược đa dạng, có hướng dẫn chơi cho từng trò chơi rõ ràng
- Giao diện website và App mobile trực quan, khoa học
- Bộ phận chăm sóc và hỗ trợ bet thủ được đào tạo bài bản, hoạt động 24/24
- Chương trình khuyến mãi cho người chơi hấp dẫn
Nhược điểm:
- Còn khá mới thị trường cá cược Việt Nam
We88 – Thưởng ngay 100K tiền cược miễn phí khi xác nhận tài khoản
Nhắc tới các nhà cái đến từ châu Âu uy tín đang có mặt tại Việt Nam thì chắc chắn không thể bỏ qua We88. Đây là một sân chơi cá cược online lớn, sở hữu kho game cá cược cực đồ sộ. Dù bạn muốn tham gia cá cược thể thao, E-thể thao hay chơi game Bắn Cá, Sòng Bài, Nổ Hũ,… nhà cái đều có thể đáp ứng.
We88 – Thưởng ngay 100K tiền cược miễn phí khi xác nhận tài khoản
Các trò chơi của We88 đều có tỷ lệ thưởng tốt. Chưa hết, nhà cái còn đưa ra nhiều chương trình khuyến mãi hấp dẫn như tặng tiền cược miễn phí, thưởng chào mừng thành viên mới,… với giá trị thưởng cao.
| Tên công ty |
Moon Technologie NV |
| Địa chỉ |
Curacao |
| Giấy phép |
Curacao eGaming |
| Chương trình VIP |
Có |
| Ứng dụng |
Có |
| Phí |
Không |
Ưu điểm:
- Nhà cái trực tuyến hợp pháp, được tập đoàn lớn điều hành
- Cung cấp website và App mobile cho bet thủ cá cược tiện lợi
- Game cá cược phong phú, chất lượng, không lo gian lận
- Nhiều hình thức nạp tiền và rút tiền với tốc độ nhanh chóng, và miễn phí
- Nhiều chương trình khuyến mãi lớn
Nhược điểm:
- Mới hoạt động tại Việt Nam nên chưa được biết tới rộng rãi
1xBet – Nhà cái châu Âu hỗ trợ cá cược bằng tiền điện tử trên website và App mobile
Cũng nằm trong danh sách top các nhà cái châu Âu uy tín và đáng chơi nhất năm, 1xBet được mệnh danh là “thiên đường” dành cho các bet thủ muốn cá cược bằng tiền điện tử. Bạn có thể sử dụng bất kỳ loại tiền điện tử nào để cá cược thể thao, cá cược Esport hay chơi game Casino, TV Game, Slot game,… tại đây.
1xBet – Cá cược dễ dàng bằng tiền điện tử trên website và App mobile
Ngoài tiền thưởng thắng cược thì cá cược tại 1xBet bạn còn có cơ hội nhận những khuyến mãi cực kỳ giá trị trong khi điều kiện, quy định khuyến mãi lại khá “cởi mở”.
| Tên công ty |
Giskard Datatech Ovt. Ltd |
| Địa chỉ |
Limassol, Cyprus |
| Giấy phép |
Curaçao eGaming License |
| Chương trình VIP |
Có |
| Ứng dụng |
Có |
| Phí |
Không |
Ưu điểm:
- Nhà cái châu Âu hợp pháp, có thâm niên hoạt động lâu năm
- Kho game cá cược đồ sộ, phong phú về thể loại
- Cá cược bằng tiền điện tử nên rất an toàn, bảo mật
- Nạp – rút tiền nhanh chóng, dễ dàng
Nhược điểm:
- Không hỗ trợ cá cược bằng tiền fiat
Instant Casino – Đăng ký tài khoản nhận thưởng chào mừng tới €7.500
Cái tên tiếp theo mà chúng tôi muốn đề xuất với bet thủ Việt đó là Instant Casino. Mặc dù chỉ mới đi vào hoạt động từ đầu năm 2024 nhưng nhà cái đã nhanh chóng đứng vững trên thị trường châu Âu, thậm chí là mở rộng phạm vi ảnh hưởng sang cả châu Á, trong đó có Việt Nam và thu hút hàng trăm nghìn lượt đăng ký tài khoản.
Instant Casino – Đăng ký tài khoản nhận thưởng chào mừng tới €7.500
Thành tựu này có được là nhờ Instant Casino đã tích cực hợp tác với những nhà phát hành game lớn như Playtech, Gameplay, NetEnt, JILI, Pragmatic Play, Evolution,… để cung cấp các game cá cược chất lượng với tỷ lệ thưởng cao cùng khuyến mãi siêu khủng.
| Tên công ty |
SIMBA N.V. |
| Địa chỉ |
Curacao |
| Giấy phép |
Curacao eGaming |
| Chương trình VIP |
Có |
| Ứng dụng |
Có |
| Phí |
Không |
Ưu điểm:
- Nhà cái online được cấp phép hoạt động hợp pháp, an toàn cho bet thủ
- Giao diện cược được thiết kế khoa học, thân thiện người dùng
- Nạp rút tiền đơn giản, tốc độ xử lý giao dịch nhanh
- Bộ phận hỗ trợ khách hàng sẵn sàng tư vấn, và hỗ trợ người chơi 24/7
- Cộng đồng bet thủ đánh giá tích cực
Nhược điểm:
- Giá trị các khuyến mãi cao nhưng các chương trình khuyến mãi chưa đa dạng
Coin Casino – Nhà cái đến từ châu Âu uy tín và chất lượng nhất
Nhắc đến các nhà cái uy tín đến từ châu Âu, CoinCasino chắc chắn là cái tên không thể bỏ qua. Đây là nền tảng cá cược trực tuyến bằng tiền điện tử được cấp phép hoạt động hợp pháp bởi tổ chức cá cược hàng đầu hiện nay.
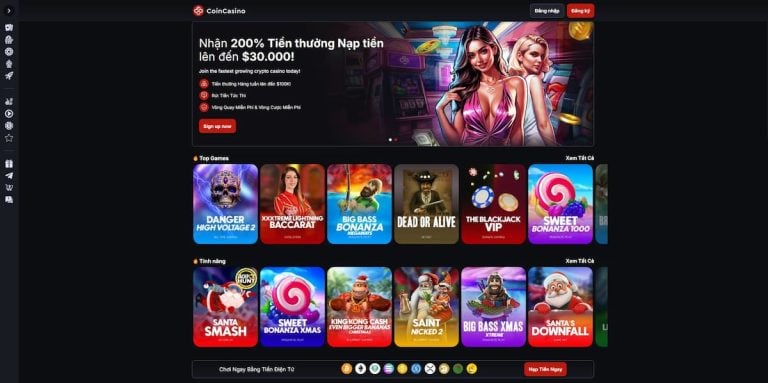
Đặc biệt, CoinCasino nâng cao tính an toàn bằng cách sử dụng Free Blocks – hệ thống bảo vệ tài sản kỹ thuật số hàng đầu thế giới – để bảo vệ tài sản của người chơi. Hệ thống này kết hợp cùng công nghệ MPC (Mật mã tính toán đa bên), giúp ngăn chặn hiệu quả các cuộc tấn công mạng và giảm thiểu rủi ro.
Không chỉ mang lại sự an tâm tuyệt đối, Coin Casino còn mở ra một thế giới giải trí đỉnh cao với hơn 4.000 trò chơi đa dạng. Từ cá cược thể thao, casino trực tiếp đến các trò chơi hiện đại như slot, crash game,… người chơi sẽ luôn tìm thấy trải nghiệm phù hợp với sở thích của mình.
| Tên công ty |
Igloo Ventures SRL |
| Địa chỉ |
San Jose, Cedula Juridica, Costa Rica |
| Giấy phép |
Chính phủ của Đảo Tự trị Anjouan |
| Chương trình VIP |
Có |
| Ứng dụng |
Không |
| Phí |
Không |
Ưu điểm:
- Nhà cái đến từ châu Âu hợp pháp, có danh tiếng tốt trên thị trường
- Nạp rút bằng tiền điện tử, hoàn toàn ẩn danh
- Thiết kế giao diện website và trên di động trực quan, dễ sử dụng
- Hệ thống bảo mật cực kỳ hiện đại, đảm bảo thông tin cũng như tài sản của người chơi
- Chăm sóc và hỗ trợ khách hàng chuyên nghiệp, 247
Nhược điểm:
- Chương trình khuyến mãi chưa đa dạng so với các đối thủ trong ngành
Tiêu chí xác định nhà cái đến từ châu Âu uy tín

Để có thể đề xuất cho bet thủ những nhà cái đến từ châu Âu thực sự uy tín, đáng tin cậy, chúng tôi đã trực tiếp trải nghiệm sản phẩm, dịch vụ cá cược tại từng nhà cái và đưa ra đánh giá dựa trên bộ tiêu chí:
- Giấy phép hoạt động: Các nhà cái đều phải được cấp phép kinh doanh bởi tổ chức quản lý dịch vụ cá cược trực tuyến thuộc chính phủ. Điều này sẽ đảm bảo tính pháp lý và sự an toàn cho bet thủ khi tham gia cá cược
- Bảo mật thông tin: Chúng tôi chỉ đề xuất nhà cái có hệ thống bảo mật hiện đại, được cấp chứng chỉ bởi tổ chức uy tín. Mục đích là để tránh cho người chơi bị rò rỉ thông tin trong quá trình tham gia cá cược
- Thiết kế giao diện: Giao diện ảnh hưởng trực tiếp tới trải nghiệm cá cược của người chơi. Do đó, trong quá trình cá cược tại các nhà cái, chúng tôi đã đánh giá cả thiết kế giao diện. Chúng tôi chỉ đề xuất nhà cái có thiết kế giao diện chuyên nghiệp, trực quan, dễ cá cược, hỗ trợ đa ngôn ngữ, bao gồm cả tiếng Việt và hoạt động ổn định trên nhiều thiết bị khác nhau
- App mobile cho Android, iOS: Ngoài ra, chúng tôi còn đánh giá các nhà cái đến từ châu Âu dựa vào ứng dụng cá cược. Người chơi ngày càng có xu hướng thích cá cược trên điện thoại. Vì vậy, nhà cái cần phải có App mobile cho Android và iOS của riêng mình để người chơi cá cược dễ dàng, thuận lợi hơn
- Phương thức thanh toán: Cần phải đa dạng, hỗ trợ nhiều phương thức với thủ tục đơn giản và không tính phí
- Chương trình khuyến mãi: Ưu tiên đề xuất nhà cái có các khuyến mãi giá trị, điều kiện nhận thưởng đơn giản, không cố tình làm khó người chơi
- Chăm sóc khách hàng: Chỉ đề xuất nhà cái có nhiều kênh liên hệ, phản hồi người chơi rõ ràng, giải đáp chi tiết với thái độ lịch sự
Các khuyến mãi của nhà cái châu Âu
Tham gia cá cược tại nhà cái đến từ châu Âu bạn sẽ có cơ hội nhận được các khuyến mãi sau:
- Khuyến mãi chào mừng thành viên mới
- Khuyến mãi nạp lại
- Thưởng hoàn trả không giới hạn
- Thưởng tiền cược miễn phí
Ngoài ra còn có rất nhiều khuyến mãi khác. Mỗi nhà cái sẽ có các chương trình khuyến mãi riêng.
Kết luận
Trên đây là danh sách nhà cái đến từ châu Âu và những đánh giá tổng quan của chúng tôi. Hãy tham gia cá cược ở nhà cái châu Âu uy tín để có trải nghiệm cá cược tuyệt vời và an toàn.
Câu hỏi thường gặp
Bao nhiêu tuổi có thể tham gia cá cược tại nhà cái đến từ châu Âu?
Các nhà cái tới từ châu Âu quy định người chơi phải đủ 18 tuổi mới có thể đăng ký tài khoản thành viên và tham gia cá cược.
Chơi gì tại nhà cái tới từ châu Âu?
Tại nhà cái tới từ châu Âu có rất nhiều trò chơi thuộc đa dạng thể loại cho bạn trải nghiệm như: Cá cược thể thao, cá cược Esport, Slot game, Xổ số, Bắn cá,…